



富士通と大阪大が量子ソフトウェア領域でまた一歩前進
創薬・新素材開発で計算を“1000倍高速化” 量子コンピューター実用化を見据えた新技術
2026年03月26日 08時00分更新

「量子コンピューター研究は総力戦」と語るのは、大阪大学の藤井啓祐教授である。量子コンピューターの実用化に向け、様々な領域で研究開発が進む中、同大学と富士通が連携するのが量子ソフトウェア開発だ。
富士通と大阪大学の量子情報・量子生命研究センターは、2026年3月25日、量子コンピューターによる「化学材料のエネルギー計算」を最適化する、新技術を開発したことを発表した。同技術を用いることで、従来手法で数千日かかる高度なエネルギー計算を最大1000分の1に縮め、今後登場する数万量子ビットの量子コンピューターの産業応用に寄与するという。
記者向けの技術説明会で、富士通の量子研究所長である佐藤信太郎氏は、市場規模の大きい化学材料分野の応用は「大きなインパクト」だと強調した。

富士通 富士通研究所フェロー 兼 量子研究所長 佐藤信太郎氏
富士通と大阪大学の“誤り耐性実用化”に向けた歩み
量子コンピューター実用化の鍵となっているのが、外部ノイズに起因する「量子エラー」をどう防ぐかにある。この解決策として期待されているのが、量子ビットを論理量子ビットに冗長化して、エラー訂正をしながら計算を進める「誤り耐性量子計算(FTQC)」の確立である。
富士通と大阪大学は、2021年に大阪大学内で共同研究部門を設置し、FTQCに向けた量子ソフトウェアの共同研究を進めてきた。その最初の成果となったのが、2023年に発表された「STARアーキテクチャ」である。
前提として、FTQCでは論理量子ビットに対する操作(基本量子ゲート)を組み合わせることで量子計算を実行する。従来アーキテクチャでは、この中の「論理Tゲート(量子ビットの位相角を45度回転させる操作)」の繰り返しにおいて、大量の量子ビットを必要とすることが課題だった。

誤り耐性量子計算(FTQC)の概要
そこでSTARアーキテクチャでは、コストの高い論理Tゲートを、位相回転操作を効率化した独自の「位相回転ゲート」に置き換えた。これにより、必要になる量子ビット数やゲート操作数を1桁以上減らすことに成功している。「ただし、位相回転ゲートはエラー訂正によって守られておらず、トレードオフの関係にある。幾分かのエラーを受け入れて、リソースを大幅に減らす試み」と佐藤氏。
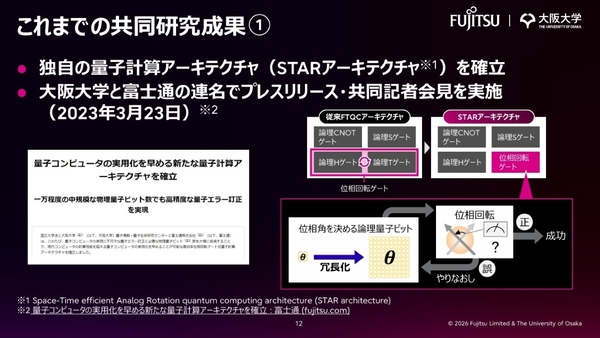
STARアーキテクチャ ver. 1
さらに2024年には、位相回転ゲートの操作精度を向上させたSTARアーキテクチャのver. 2を発表。これにより計算規模を1000倍に拡大させ、その結果、数万量子ビットの量子コンピューターを用いた固体材料の物性計算を現実化した。
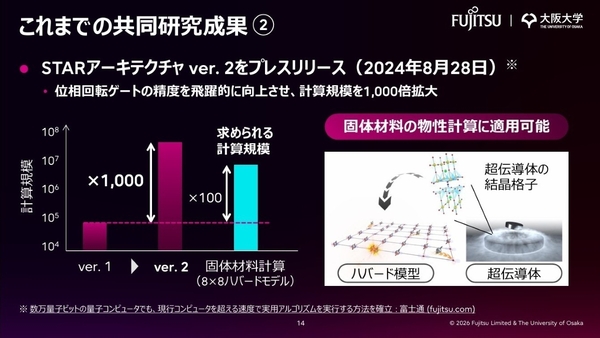
STARアーキテクチャ ver. 2
“科学材料の物性計算”を現実化する2つの新技術
そして、今回発表されたのが、STARアーキテクチャのさらなる精度向上と分子モデル(科学材料の計算対象)を最適化する新技術である。
今回の研究の目的は、複雑な構造を持つ科学材料の分子エネルギーの計算を最適化して、将来の量子コンピューターの応用範囲を「科学材料の物性計算」にまで広げることにある。
STARアーキテクチャのver. 2で対応した固定材料に比べ、化学材料は大規模な量子ビット数が求められるものの、市場規模が桁違いに大きい。固定材料の対象である超伝導材の市場が93億ドルに対し、化学材料の対象である「製薬・創薬」「アンモニア合成」「カーボンリサイクル」の市場合算は、その200倍以上に上る。

化学材料分野の市場規模
まず、STARアーキテクチャのver.3では、計算精度を約10倍向上させ、同じ量子ビット数での計算規模を拡大している。これにより、量子コンピューターに要求される物理エラー率も緩和可能だ。具体的には、ver. 1、2で置き換えた独自の位相回転ゲートに、一度はなくした論理Hゲートを融合させることで実現した。
これは、位相回転ゲートにおける位相回転の連続失敗が、計算精度を悪化させる要因だと判明したからだ。そこで、一定の回転失敗が続くと、論理Hゲートによる位相回転に切り替える仕組みを採用。こうして、適切に位相回転ゲートと論理Tゲートを組み合わせることで、リソースと精度の問題を同時に解決した。
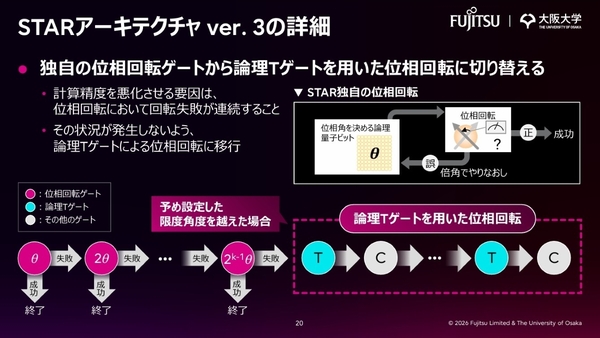
STARアーキテクチャ Ver3
ただ、「STARアーキテクチャの改良だけでは、分子モデルのシミュレーションには届かない」と佐藤氏。そこで開発されたのが、分子モデルの最適化技術である。
科学材料の分子エネルギーの計算には、分子モデルを構築して、そこから量子回路を生成し、量子コンピューターで計算するという手順を踏む。この電子回路は分子モデルを複数の項に分割して生成される。その際、項の数に応じて高コストな「時間発展法」と項の重要度に応じて高コストになる「ランダムサンプリング法」という、2つの計算手法を組み合わせることで計算時間(コスト)が削減できる。
今回、近似精度を維持したまま分子モデルを変形させる新技術により、重要度の分布を変え、2つの計算手法のバランスを最適化した。これにより、分子エネルギーの計算時間を大幅に削減している。
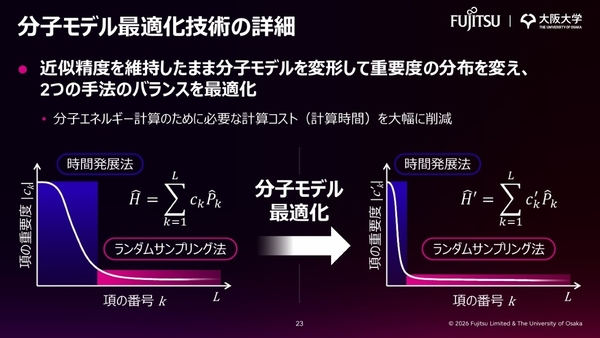
分子モデル最適化技術
本記事はアフィリエイトプログラムによる収益を得ている場合があります



